









Contents
Wilson’s disease
What is it ?
Wilson’s disease is an inherited genetic disease that prevents the elimination of copper from the body. The buildup of copper in the liver and brain causes liver or neurological problems. The prevalence of Wilson’s disease is very low, around 1 in 30 people. (000) There is an effective treatment for this disease, but its early diagnosis is problematic because it remains silent for a long time.
Symptoms
Copper build-up begins at birth, but the first symptoms of Wilson’s disease often do not appear until adolescence or adulthood. They can be very diverse because several organs are affected by the accumulation of copper: the heart, kidneys, eyes, blood… The first signs are hepatic or neurological in three-quarters of cases (40% and 35% respectively) , but they can also be psychiatric, renal, hematological and endocrinological. The liver and brain are particularly affected because they already naturally contain the most copper. (2)
- Liver disorders: jaundice, cirrhosis, liver failure …
- Neurological disorders: depression, behavioral disorders, learning difficulties, difficulties in expressing themselves, tremors, cramps and contractures (dystonia) …
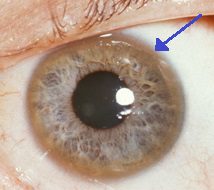
The Keyser-Fleisher ring that surrounds the iris is characteristic of the buildup of copper in the eye. In addition to these acute symptoms, Wilson’s disease can present with uncharacteristic symptoms, such as general fatigue, abdominal pain, vomiting and weight loss, anemia, and joint pain.
The origins of the disease
At the origin of Wilson’s disease, there is a mutation in the ATP7B gene located on chromosome 13, which is involved in the metabolism of copper. It controls the production of an ATPase 2 protein which plays a role in transporting copper from the liver to other parts of the body. Copper is a necessary building block for many cell functions, but in excess of copper it becomes toxic and damages tissues and organs.
Risk factors
Transmission of Wilson’s disease is autosomal recessive. It is therefore necessary to receive two copies of the mutated gene (from the father and the mother) to develop the disease. This means that men and women are equally exposed and that two parents carrying the mutated gene but not sick have a risk in four at each birth of transmitting the disease.
Prevention and treatment
There is an effective treatment to stop the progression of the disease and reduce or even eliminate its symptoms. It is also necessary that it be initiated early, but it often takes many months after the onset of symptoms to diagnose this silent disease, little known and whose symptoms point to many other conditions (hepatitis for which is liver damage and depression for psychiatric involvement).
A “chelating” treatment makes it possible to attract copper and eliminate it in the urine, thus limiting its accumulation in the organs. It is based on D-penicillamine or Trientine, medicines taken by mouth. They are effective, but can cause serious side effects (kidney damage, allergic reactions, etc.). When these side effects are too important, we resort to the administration of zinc which will limit the absorption of copper by the intestines.
A liver transplant may be necessary when the liver is too damaged, which is the case for 5% of people with Wilson disease (1).
A genetic screening test is offered to the siblings of an affected person. It gives rise to an effective preventive treatment in the event that a genetic abnormality is detected in the ATP7B gene.