









Contents
Bourneville tuberous sclerosis
What is it ?
Bourneville tuberous sclerosis is a complex genetic disease characterized by the development of a benign (non-cancerous) tumor in different parts of the body. These tumors can then be located in the skin, brain, kidneys, and other organs and tissues. This pathology can also cause serious problems in the development of the individual. However, the clinical manifestations and severity of the disease vary from patient to patient.
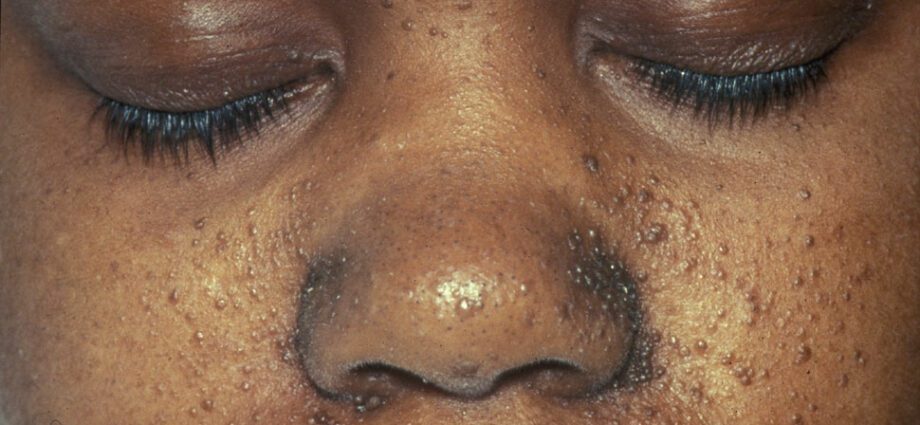
The associated skin abnormalities are generally similar to spots on the skin or to areas where the skin is lighter than on the rest of the body. The development of tumors in the face is called angiofibroma.
In the context of brain damage, the clinical signs are epileptic seizures, behavioral problems (hyperactivity, aggressiveness, intellectual disabilities, learning problems, etc.). Some children with the disease even have some form of autism, developmental disorders, affecting social interactions and communication. Benign brain tumors can also cause complications that can be fatal to the subject.
The development of tumors in the kidneys is common in people with tuberous sclerosis. This can cause severe complications in kidney function. In addition, tumors can develop in the heart, lungs and retina. (2)
It is a rare disease, the prevalence of which (number of cases in a given population at a given time) amounts to 1 / 8 to 000 / 1 people. (15)
Symptoms
The clinical manifestations associated with tuberous sclerosis of Bourneville vary according to the organs affected. In addition, the symptoms associated with the disease vary widely from one individual to another. With symptoms ranging from mild to severe.
The most widely identified symptoms of this disease include epileptic seizures, cognitive and behavioral disorders, skin abnormalities, etc. The organs most often affected are: the brain, the heart, the kidneys, the lungs and the skin.
The development of malignant (cancerous) tumors is possible in this disease but are rare and mainly affect the kidneys.
The clinical signs of the disease in the brain originate from attacks at different levels:
– damage to cortical tubercles;
– ependymal nodules (SEN);
– giant ependymal astrocytomas.
They result in: the development of mental retardation, learning difficulties, behavioral disorders, aggressiveness, attention disorders, hyperactivity, obsessive-compulsive disorders, etc.
Kidney damage is characterized by the development of cysts or angiomyolipomas. These can lead to kidney pain and even kidney failure. If heavy bleeding is noticeable, it may be from severe anemia or high blood pressure. Other more serious but rare consequences may also be visible, in particular the development of carcinomas (tumor of the constituent cells of the epithelium).
Eye damage can be similar to visible spots on the retina, causing visual disturbances or even blindness.
Skin abnormalities are numerous:
– hypomelanic macules: which result in the appearance of light spots on the skin, anywhere on the body, as a result of a deficiency in melanin, a protein that gives color to the skin;
– the appearance of red spots on the face;
– discolored patches on the forehead;
– other skin abnormalities, dependent from one individual to another.
Lung lesions are present in 1/3 of patients with a slight female predominance. The associated symptoms are then more or less severe breathing difficulties.
The origins of the disease
The origin of the disease is genetic and hereditary.
Transmission involves mutations in the TSC1 and TSC2 genes. These genes of interest come into play in the formation of proteins: hamartin and tuberin. These two proteins make it possible, through an interactive game, to regulate cell proliferation.
Patients with the disease are born with at least one mutated copy of these genes in each of their cells. These mutations then limit the formation of hamartine or tubertine.
In the context where the two copies of the gene are mutated, they completely prevent the production of these two proteins. This protein deficiency therefore no longer allows the body to regulate the growth of certain cells and, in this sense, leads to the development of tumor cells in different tissues and / or organs.
Risk factors
The risk factors for developing such a pathology are genetic.
Indeed, the transmission of the disease is effective through an autosomal dominant mode. Either, the mutated gene of interest is located at a non-sexual chromosome. In addition, the presence of only one of the two copies of the mutated gene is sufficient for the disease to develop.
In this sense, an individual possessing one of these two parents suffering from the disease has a 50% risk of developing the sick phenotype himself.
Prevention and treatment
The diagnosis of the disease is first of all differential. It is based on atypical physical criteria. In most cases, the first characteristic signs of the disease are: the presence of recurrent epileptic seizures and delays in the subject’s development. In other cases, these first signs result in skin spots or the identification of a heart tumor.
Following this first diagnosis, additional examinations are essential in order to validate the diagnosis or not. These include:
– a brain scan;
– an MRI (Magnetic Resonance Imaging) of the brain;
– an ultrasound of the heart, liver and kidneys.
The diagnosis can be effective at the birth of the child. Otherwise, it is important that it be carried out as quickly as possible in order to take charge of the patient as soon as possible.
Currently, there is no cure for the disease. The associated treatments are therefore independent of the symptoms presented by each individual.
Usually, anti-epileptic drugs are given to limit seizures. In addition, drugs for the treatment of tumor cells of the brain and kidneys are also prescribed. In the context of behavioral problems, specific treatment of the child is necessary.
Treatment of the disease is usually long term. (1)