









Contents
Huntington’s disease
What is it ?
Huntington’s disease is a genetic and inherited neurodegenerative disease. By destroying neurons in certain areas of the brain, it causes severe motor and psychiatric disorders and can lead to complete loss of autonomy and death. The gene whose alteration causes the disease was identified in the 90s, but Huntington’s disease remains incurable to this day. It affects one in 10 people in France, which represents around 000 patients.
Symptoms
It is still sometimes called “Huntington’s chorea” because the most characteristic symptom of the disease are the involuntary movements (called choreic) it causes. However, some patients do not present with choreic disorders and the symptoms of the disease are broader: to these psychomotor disorders are frequently added psychiatric and behavioral disorders. These psychiatric disorders which occur frequently at the onset of the disease (and sometimes appear before the motor disorders) can lead to dementia and suicide. Symptoms usually appear around 40-50 years of age, but early and late forms of the disease are observed. Note that all carriers of the mutated gene one day declare the disease.
The origins of the disease
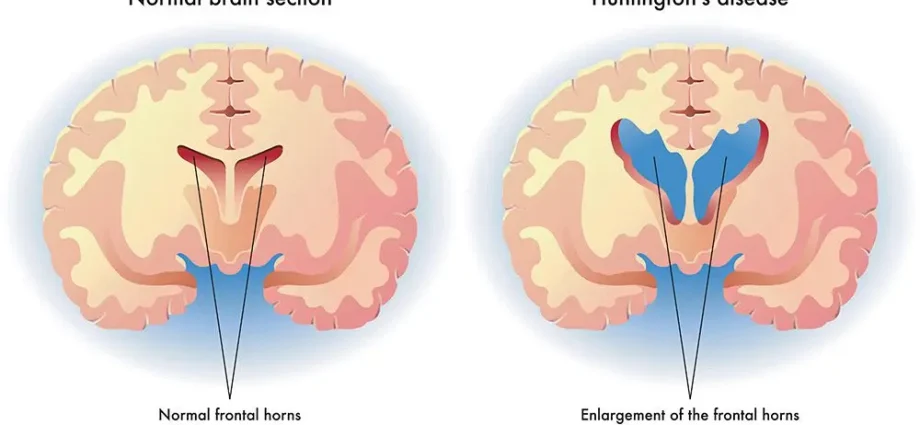
American physician George Huntington described Huntington’s disease in 1872, but it wasn’t until 1993 that the responsible gene was identified. It was localized on the short arm of chromosome 4 and named IT15. The disease is caused by a mutation in this gene that controls the production of the huntingtin protein. The precise function of this protein is still unknown, but we know that the genetic mutation makes it toxic: it causes deposits in the middle of the brain, more precisely in the nucleus of neurons of the caudate nucleus, then of the cerebral cortex. However, it should be noted that Huntington’s disease is not systematically linked to IT15 and can be caused by mutating other genes. (1)
Risk factors
Huntington’s disease can be passed from generation to generation (it is called “autosomal dominant”) and the risk of transmission to the offspring is one in two.
Prevention and treatment
The genetic screening of the disease in people at risk (with a family history) is possible, but very supervised by the medical profession, because the result of the test is not without psychological consequences.
Prenatal diagnosis is also possible, but it is strictly framed by law, because it raises questions of bioethics. However, a mother who is considering a voluntary termination of pregnancy in the event that her fetus carries the altered gene has the right to request this prenatal diagnosis.
To date, there is no curative treatment and only the treatment of symptoms can relieve the sick person and slow down their physical and psychological deterioration: psychotropic drugs to alleviate the psychiatric disorders and the episodes of depression which often go hand in hand with the disease. ; neuroleptic drugs to reduce choreic movements; rehabilitation through physiotherapy and speech therapy.
The search for future therapies is directed towards the transplant of fetal neurons in order to stabilize the motor functions of the brain. In 2008, researchers from the Pasteur Institute and the CNRS proved the brain’s ability to self-repair by identifying a new source of neuron production. This discovery raises new hopes for the treatment of Huntington’s disease and other neurodegenerative conditions, such as Parkinson’s disease. (2)
Gene therapy trials are also underway in several countries and are moving in several directions, one of which is to block the expression of the mutated huntingtin gene.